Amyloïdose cardiologie
Cardiale betrokkenheid bij amyloïdose
Als amyloïdfibrillen het hart infiltreren
Epidemiologie | Symptomen | Diagnose | Behandeling
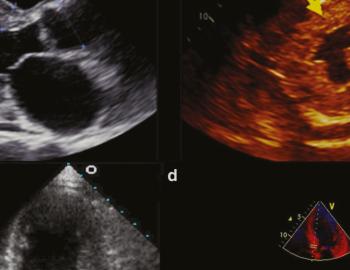
Een patiënt met cardiale amyloïdose heeft een hart waarvan de extracellulaire ruimte van het myocard is geïnfiltreerd door amyloïdfibrillen. Het gevolg? Cardiomyopathie met ernstige klachten en een sombere prognose. De meeste patiënten hebben niet lang meer te leven en overlijden door hartfalen of plotse hartdood.1 Een vroege diagnose kan dat veranderen.2
Gissen geblazen: de epidemiologie van cardiale amyloïdose
De epidemiologie van amyloïdose in kaart brengen is een uitdaging. Waarom is dat zo? Dat heeft onder andere met onderdiagnose te maken. Amyloïdosepatiënten presenteren zich vaak met uiteenlopende, aspecifieke symptomen. Daarom wordt het ontstaan van deze symptomen nogal eens aan een andere aandoening toegeschreven.
Zo kan een patiënt gediagnosticeerd zijn met (onbegrepen) hartfalen. Terwijl diegene eigenlijk amyloïdose met cardiale betrokkenheid heeft. Door zulke gemiste diagnoses bestaat het risico dat de cijfers onderschatten hoe vaak de ziekte voorkomt.
Bovendien komt cardiale betrokkenheid vaker voor bij de ene vorm van amyloïdose dan bij de andere. Ook dat maakt het berekenen van eenduidige epidemiologische parameters lastig.
Toch zijn er cijfers beschikbaar. AL amyloïdose is de vorm die wereldwijd het meest voorkomt en in Nederland een incidentie heeft van ongeveer 10 patiënten per miljoen inwoners per jaar. Tot wel 80% van hen presenteert zich met cardiale uitingen van de ziekte.3
Prevalentie van ATTR cardiale amyloïdose wordt geschat tot op 13% bij HFpEF4, 16% bij TAVI5
De prevalentie van ATTR-CM is lastig met harde cijfers te onderbouwen door onderdiagnose. Wat we weten op basis van literatuur is dat wtATTR-CM voorkomt bij 5-13% van de HFpEF patiënten.4 De Nederlandse HF populatie is 242.300 (2018), waarvan 113.881 HFpEF (47%).
Op basis van de bovenstaande gegevens kunnen we stellen dat amyloïdose een zeer zeldzame aandoening is. Maar zodra we die ziekte in het licht van hartfalen bekijken, wordt duidelijk dat amyloïdose een niet te onderschatten rol speelt binnen de cardiologie.
Over de prevalentie van de verworven variant van ATTR-amyloïdose is nog weinig bekend. Een studie onder patiënten met een behouden ejectiefractie liet een prevalentie van 13% zien.1
De symptomen van cardiale amyloïdose
Bij een aantal vormen van amyloïdose kan het hart betrokken zijn. ATTR amyloïdose is daar een voorbeeld van.
Amyloïdosepatiënten met cardiale betrokkenheid krijgen op den duur last van hun hart, omdat amyloïdfibrillen zich daar opstapelen en schade veroorzaken. Schade aan onder andere de atria, de ventrikels en de perivasculaire ruimte. Soms raken ook de hartkleppen en het geleidingssysteem van het hart aangetast.
Hoe ziet zo’n beschadigd hart eruit?
Cardiale amyloïdose leidt tot een progressieve toename in de dikte van het myocard; mogelijk zowel van het rechterventrikel als van het linkerventrikel. Hetzelfde geldt voor het interatriale septum en de atrioventriculaire kleppen.7 Het resultaat is hartfalen, vaak met behouden ejectiefractie (HFpEF). De meest voorkomende symptomen die daarbij horen, zijn:
- vermoeidheid;
- dyspneu;
- oedeem.
Verder kan een patiënt met cardiale amyloïdose zich presenteren met symptomen zoals: aritmiën in de vorm van ventriculaire tachycardie, geleidingsstoornissen en pompfunctiestoornissen.8
Patiënten met AL amyloïdose en ATTR amyloïdose kunnen ook een aantal andere symptomen krijgen die niets met het hart te maken hebben. In dat geval hopen de amyloïdfibrillen zich niet alleen op in het hart, maar ook in andere organen en weefsels.
Dat gebeurt bijvoorbeeld bij de wildtype vorm van ATTR, waar amyloïdinfiltratie het ligament van de carpale tunnel beschadigt. Dat zorgt voor een tintelend of pijnlijk gevoel in handen en vingers, veroorzaakt door het carpaletunnel-syndroom. Meestal presenteren deze patiënten zich met dit klachtenpatroon in een vroeg stadium. Dat wil zeggen: jaren voordat de cardiale symptomen zichtbaar worden. Tot wel 50% van de patiënten met wildtype ATTR heeft een voorgeschiedenis van bilateraal carpaletunnel-syndroom.9
Ook bij erfelijke ATTR amyloïdose kunnen extracardiale verschijnselen optreden die typerend zijn voor de ziekte. Bijvoorbeeld polyneuropathie (met of zonder autonome disfunctie), diarree, obstipatie en oogproblemen.
De bovengenoemde symptomen zijn eveneens kenmerkend voor AL amyloïdose. Daarnaast is macroglossie een klinisch verschijnsel dat specifiek is voor deze vorm van amyloïdose.
Bent u op zoek naar de oorzaak van hartfalen bij uw patiënt? Of heeft u het vermoeden dat er sprake is van ATTR-CM als onderliggend lijden? Bekijk onderstaand schema met de belangrijkste aanwijzingen die in verband zijn gebracht met deze aandoening.
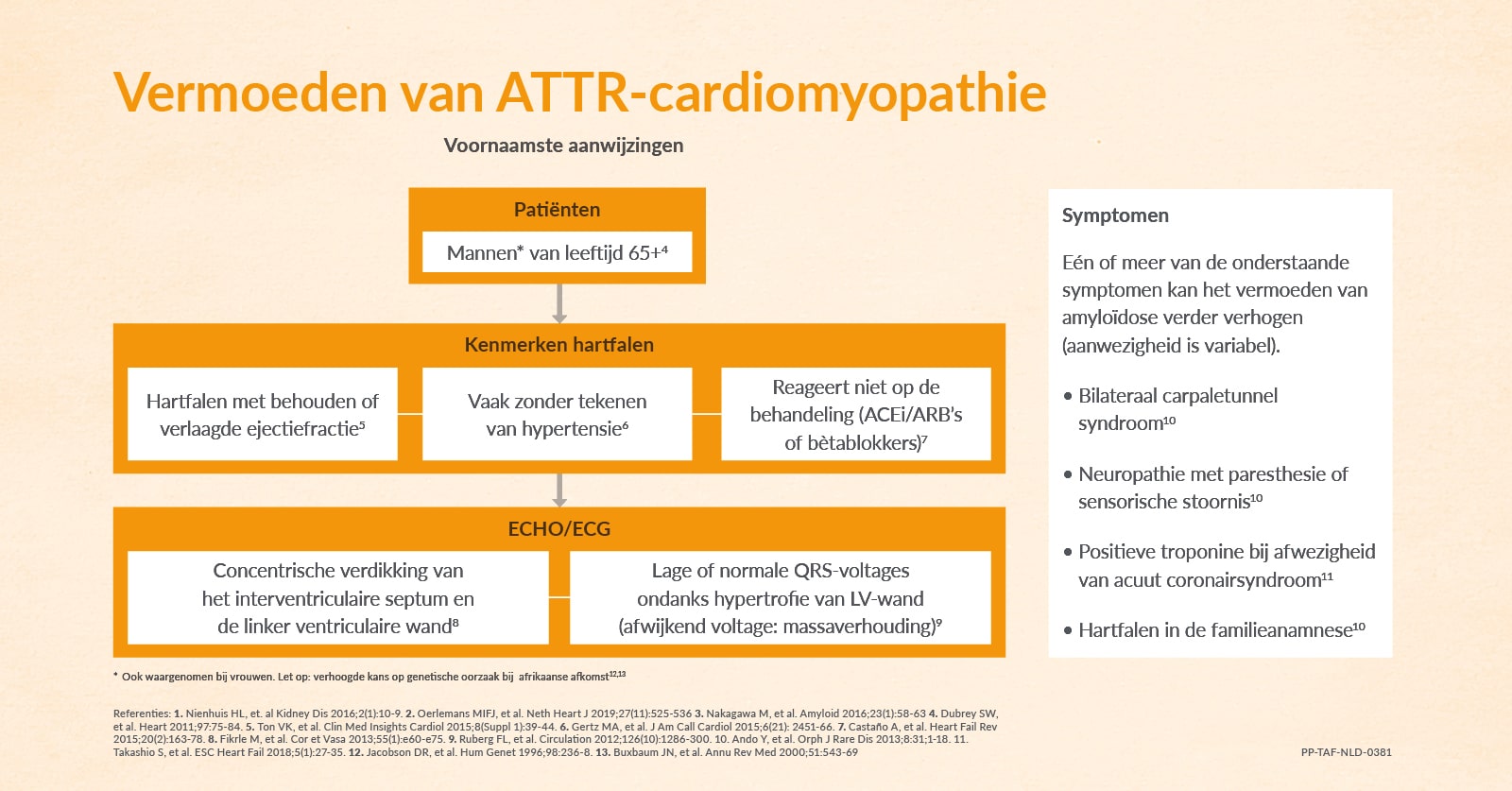
Behoefte aan meer achtergrondinformatie? Via de onderstaande link downloadt u het overzicht met de klinische symptomen die kunnen wijzen op cardiale amyloïdose in het algemeen en ATTR-CM in het bijzonder.
Cardiale amyloïdose wordt vaak verkeerd gediagnosticeerd. Of het wordt te laat herkend door een laag ziektebewustzijn en andere ziektegerelateerde factoren.6
Ná diagnose ligt de mediane overleving van ATTR-CM tussen de 2 en 6 jaar, afhankelijk van het genotype en de vorm.6,22 Toch is het de bittere realiteit dat die diagnose is vertraagd: gemiddeld met 3 tot 4 jaar.21
Patiënten die lijden aan AL amyloïdose met cardiale betrokkenheid hebben een nóg kortere levensverwachting. Onbehandeld is de overleving niet meer dan 6 maanden.24 Ter vergelijking: patiënten met veel voorkomende vormen van kanker of hartfalen hebben een hogere levensverwachting dan deze patiënten.8
Bij een late diagnose is er sprake van ongehinderde ziekteprogressie. Want het leidt tot uitstel van een passende behandeling. Zonder interventie blijven de amyloïdfibrillen zich opstapelen en loopt de cardiale schade op waardoor de prognose verslechtert. Daarom is een vroegtijdige diagnose essentieel.6
Om dat makkelijker voor u te maken hebben we een overzicht gemaakt van de belangrijkste symptomen die in verband zijn gebracht met cardiale amyloïdose. Inclusief ezelsbrug in de vorm van het acroniem ‘HIDDEN’. In de onderstaande animatie wordt in minder dan een minuut verteld welke symptomen dat zijn.
De behandeling van cardiale amyloïdose
Op dit moment wordt er veel onderzoek gedaan naar nieuwe en betere behandelmogelijkheden voor patienten met ATTR-amyloïdose. Meestal wordt cardiale amyloïdose op 2 manieren aangepakt. Dat gebeurt tegelijkertijd. De ene manier richt zich op congestief hartfalen. De andere manier is bedoeld om het ziekteproces van amyloïdose te vertragen door verdere afzetting van amyloïdfibrillen te voorkomen.7
Hartfalentherapie24, 26, 27
De basis van therapie voor hartfalen bestaat uit een verminderde zoutinname en farmacotherapie met diuretica en aldosteronantagonisten. Maar geen bètablokkers.
Waarom?
Die kunnen de hypotensie verergeren en de myocardiale contractiliteit verlagen, omdat ze een negatief inotroop effect hebben. Ook digoxine en calciumantagonisten zijn gecontra-indiceerd. Implanteerbare cardio-defibrillatoren en pacemakers zijn daarentegen wel geïndiceerd.
De behandeling
ATTR amyloïdose
Voor deze aandoening is de behandeling gericht op het transthyretine-eiwit (TTR) dat zich verkeerd vouwt en opstapelt. Dat gebeurt met een transthyretine stabilisator.23
Deze stabilisator bindt zich selectief en met hoge affiniteit aan het transthyretine-tetrameer. Daardoor dissocieert TTR minder snel in monomeren. En dat draagt bij aan de stabiliteit van het eiwit. Die dissociatie is de snelheidsbepalende stap van het proces dat leidt tot de vorming van amyloïd. Dus daar ingrijpen vormt de rationale voor het gebruik van stabilisators bij patiënten met ATTR-CM.29
Voor erfelijke ATTR amyloïdose bestaat nog een behandeloptie: gene-silencing. Voor de patiënten die polyneuropathie als klacht hebben, kan dit ook een behandeloptie zijn. Gene silencing is een manier om de activiteit van genen te reguleren. In dit geval is het de bedoeling om het gen te deactiveren dat verantwoordelijk is voor de productie van transthyretine.27
AL amyloïdose
Chemotherapie is de standaardbehandeling van AL amyloïdose. Het regime wordt afgestemd op de mate waarin verschillende organen zijn betrokken bij de aandoening.
Het doel van de therapie? De afwijkende plasmacelkloon uitschakelen. Daarnaast krijgt de patiënt ondersteunende behandelingen om de bijwerkingen van de chemotherapie te bedwingen.
Het inzetten van chemotherapie is effectief gebleken in het terugdringen van de verkeerd gevouwen eiwitten (het verlagen van vrije lichte ketens), maar heeft alleen nut bij deze vorm van amyloïdose.6,28
Wilt u meer weten over de behandeling van amyloïdose? Ga naar de website van het Expertisecentrum Amyloïdose (onderdeel van het UMCG). Of neem direct contact op met dit expertise centrum of dat van het UMC Utrecht.


Volg de nascholing

Skillslab Cardiale Amyloïdose
Leer interactief alles over de rode vlaggen, de presentatie en verwijsmogelijkheden. Na afloop ben je in staat patiënten vroeger te diagnosticeren en te verwijzen.
LVH Spreekuur Innovatieve ontwikkelingen in beeldvorming
Echo, ECG, MRI en PET-CT, er is een breed scala aan beeldvormende technieken beschikbaar. Wat zijn de mogelijkheden van deze technieken en wanneer zet je welke in?
Bronnen
1. Connors L, et al. Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation. 2016 Jan; 19;133(3):282-90
2. Mohty D, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013 Oct;106(10):528-40
3. Merlini G. CyBorD: stellar response rates in AL amyloidosis. Blood. 2012;119: 4343–45
4. González-López E, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–94
5. Castano A, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38:2879–87
6. Maurer M, et al. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis Circulation. 2017;135:1357–77
7. Fikrle M, et al. Cardiac amyloidosis: A comprehensive review. Cor et Vasa. 2013;55(1):e60-e75
8. Rapezzi C, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-124
9. Nakagawa M, et al. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid. 2016;23(1):58-63
10. Mohammed S, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113-122
11. Siddiqi O, et al. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28:10-21
12. Narotsky D, et al. Wild-type transthyretin cardiac amyloidosis: novel insights from advanced imaging. Can J Cardiol. 2016;32(9):1166.e1-1166.e10
13. Oerlemans, M, et al. Cardiac amyloidosis: the need for early diagnosis. Neth Heart J. 2019;27, 525–536
14. Cyrille N, et al. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol. 2014;114(7):1089-1093
15. Rapezzi C, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203-1212
16. Kumar S, at. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30:989–95.
17. Gillmore J, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39:2799–806.
18. Van den Wyngaert T, et al. The EANM practice guidelines for bone scintigraphy. Eur J Nucl Med Mol Imaging. 2016;43:1723–38
19. Gillmore J, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–12
20. Dorbala S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol. doi:10.1007 /s12350-019-01760-6
21. Arbustini E, et al. Early identification of transthyretin-related hereditary cardiac amyloidosis. J Am Coll Cardiol. 2014;7:511–4
22. Papoutsidakis N, et al. Time course of common clinical manifestations in patients with transthyretin cardiac amyloidosis: delay from symptom onset to diagnosis. J Card Fail. 2018 Feb;24(2):131-133
22. Donnelly J, et al. Cardiac amyloidosis: an update on diagnosis and treatment Clin J Med. 2017;84(12 Suppl 3):12–26
23. Castaño A, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-178
24. Sperry BW, et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol. 2018;72(17):2040-2050
25. Dubrey SW, et al. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97:75-84
26. Nativi-Nicolau J, et al. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol. 2018 Sep;33(5):571-579
27. Milani P, et al. Light Chain Amyloidosis Mediterr J Hematol Infect Dis. 2018;10(1):e2018022
28. Johnson SM, et al. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012 Aug 10;421(2-3):185-203
Ook interessant
-
De diagnose van cardiale amyloïdose
U heeft verschillende diagnostische instrumenten tot uw beschikking om cardiale betrokkenheid bij amyloïdose te bevestigen. Bij voorkeur bestaat de diagnostiek uit meerdere van deze onderzoeken, zodat u de testuitslagen uiteindelijk kunt combineren voor een definitieve diagnose.1,2
-
Wat is de prevalentie van cardiale amyloïdose binnen de regio van je ziekenhuis
Schattingen geven aan dat amyloïdose bij 10-20% van de hartfalen patienten de oorzaak van hun symptomen kunnen zijn.1 Het opnemen van het ziektebeeld in de differentiaaldiagnostiek is aan te raden. De incidentie van wild-type ATTR amyloïdose wordt geschat op 2 patiënten per miljoen inwoners per jaar. Het is niet duidelijk hoeveel van hen cardiale symptomen ontwikkelt. Op basis van deze gegevens kunnen we stellen dat amyloïdose een zeldzame aandoening is. Echter, zodra we de ziekte in het licht van hartfalen bekijken, wordt duidelijk dat amyloïdose een niet te onderschatten rol speelt binnen de cardiologie.
-
Podcasts over Amyloïdose
Amyloïdose is de naam van een groep stapelingsziekten van verkeerd gevouwen eiwitten die zich kunnen manifesteren in allerlei organen zoals hart, nieren, lever, maag-darmkanaal en het zenuwstelsel. Beluister hier verschillende podcasts over dit ziektebeeld met fascinerende gesprekken, verhalen en interessante inzichten.