
De diagnose van cardiale amyloïdose
U heeft verschillende diagnostische instrumenten tot uw beschikking om cardiale betrokkenheid bij amyloïdose te bevestigen. Bij voorkeur bestaat de diagnostiek uit meerdere van deze onderzoeken, zodat u de testuitslagen uiteindelijk kunt combineren voor een definitieve diagnose.1,2
De belangrijkste testen voor de diagnose van cardiale amyloïdose zijn:1,2
- echocardiografie en een ECG;
- geavanceerde beeldvormingstechnieken zoals een MRI-scan en botscintigrafie;
- laboratoriumonderzoek;
- genetische testen.
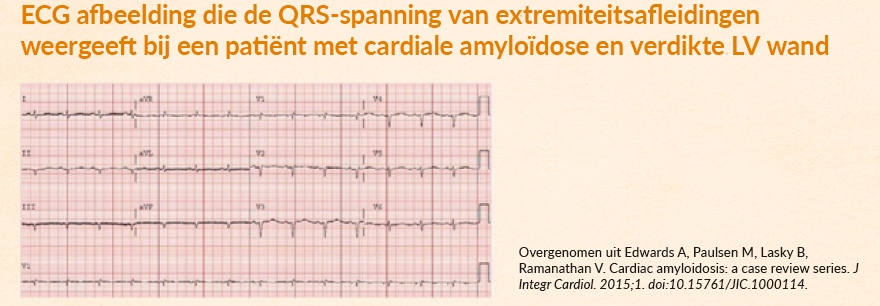
Het ECG
Een klassiek ECG-kenmerk van cardiale amyloïdose is een laag of normaal QRS-voltage, ondanks hypertrofie van het linkerventrikel.3
Hoe is dit te verklaren?
Een verdikte wand is het gevolg van zich opstapelende amyloïdfibrillen, en niet van vergrootte hartspiercellen.3
Let op, bij cardiale amyloïdose is niet altijd sprake van de ‘klassieke’ lage QRS-voltages. Bij cardiale amyloïdose zien we een discrepantie tussen de toegenomen dikte van het myocard op een echo en de omvang van de voltages op het ECG. Het is daarom van belang om niet alleen naar lage voltages te zoeken. Waarom? Dit ECG-kenmerk is pas in een laat stadium van de ziekte waar te nemen. En dat maakt het wellicht minder nuttig om cardiale betrokkenheid snel te diagnosticeren.4
De echo
Bij cardiale amyloïdose is extracellulaire infiltratie van amyloïdfibrillen verantwoordelijk voor een verdikte linkerventrikelwand (> 15 mm). Die wand is bij ATTR amyloïdose vaak dikker dan bij AL amyloïdose. Maar dat is niet altijd zo. De mate van verdikking is namelijk niet alleen afhankelijk van het type amyloïdose, maar ook van het stadium van de ziekte waarin de patiënt zich bevindt.
Dus, ziet u op de echo dat de wanddikte van het hart van uw patiënt is toegenomen, terwijl daar geen duidelijke oorzaak voor is? Dan kan dat wijzen op cardiale amyloïdose.
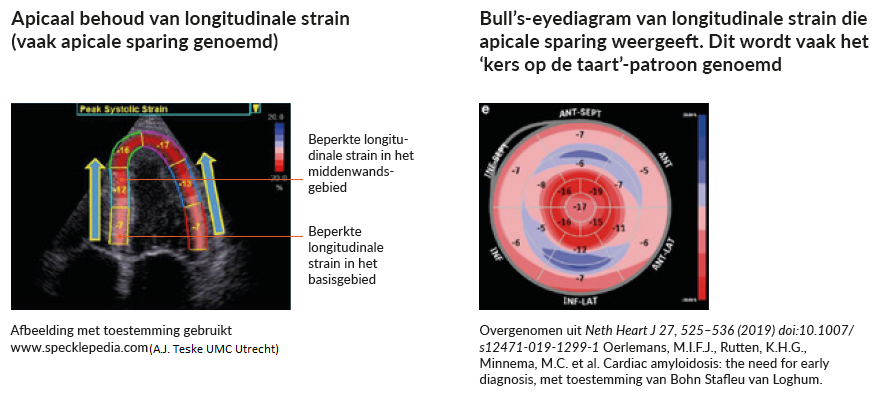
Verminderde longitudinale strain is een andere aanwijzing op de echo die het vermoeden dat uw patiënt cardiale amyloïdose heeft, kan doen toenemen.
Hierbij gaat het om een longitudinale strain die verminderd is in het basis- en middenwandsgebied. Terwijl de strain in de apex vaak wordt gespaard of behouden blijft en kan resulteren in de kenmerkende ‘kers op de taart’, zoals u in onderstaande figuur ziet.3,5,6
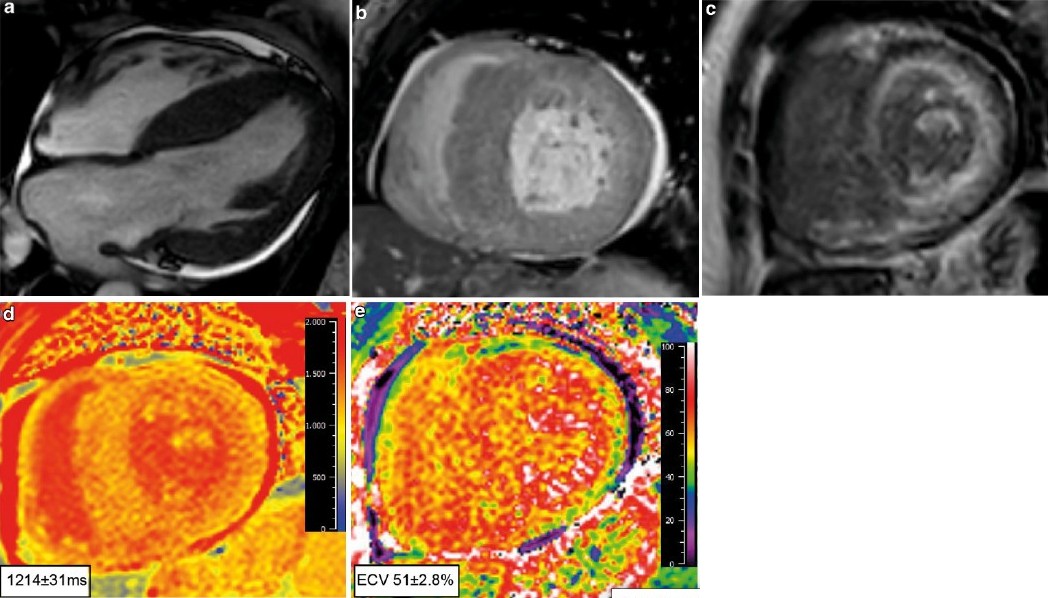
De MRI
Ook een MRI kan u specifieke kenmerken voor cardiale amyloïdose tonen, bijvoorbeeld:
- transmurale of subendocardiale late gadolinium enhancement beeldvorming (LGE);
- diffuus atriaal LGE;
- rechterventrikel LGE;
- suboptimale nulstelling als gevolg van veranderde gadoliniumkinetiek;
- verhoogd extracellulair volume en verhoogde native T1-waardes.3
Het labonderzoek
Verder is bloedonderzoek in het laboratorium noodzakelijk. Op basis van dat resultaat kunt u de diagnose AL amyloïdose snel bevestigen of verwerpen. De laborant onderzoekt serum en urine van de patiënt op de aanwezigheid van monoklonaal eiwit. Als u twijfelt over de betekenis van de uitslag, bijvoorbeeld als één van de drie labwaarden positief is, dan is overleg met de hematoloog aan te raden.3
Daarnaast zijn verhoogde waarden van NT-proBNP en troponine aanwijzingen voor cardiale betrokkenheid.7,8 Ten eerste is het NT-proBNP niveau vaak onevenredig verhoogd. Ten tweede is troponine chronisch verhoogd zonder stijging of daling. Beide bepalingen hebben zowel voor AL amyloïdose als ATTR amyloïdose een prognostische betekenis.7,8
De botscan
Botscintigrafie is een niet-invasieve manier om de diagnose ATTR amyloïdose te bevestigen, in tegenstelling tot een hartbiopt. Bovendien is het een zeer gevoelige diagnostische beeldvormingstechniek waarmee u de verschillende vormen van amyloïdose van elkaar kunt onderscheiden.9,10
Er zijn verschillende radiotracers die een sterke affiniteit met amyloïd hebben en dus geschikt zijn voor de diagnose van amyloïdose. Bijvoorbeeld: 99mTc-PYP, 99mTc-DPD en 99mTc-HMDP.11
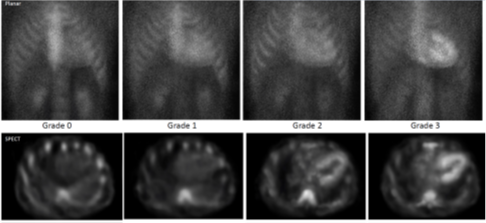
De radiotracers zijn vooral gevoelig en specifiek voor amyloïdfibrillen die het hart hebben geïnfiltreerd. Cardiale lokalisatie van een radiotracer is mogelijk bij ATTR amyloïdose en, in mindere mate, bij AL amyloïdose (zoals graad 1 traceropname, zie hieronder)..11,12
Wat is nodig om de diffuse opname van de radiotracer in het myocard te bevestigen? In ieder geval een evaluatie van de planar- en de SPECT-beelden, als onderdeel van de visuele interpretatie van de botscan.11
Als de opname van de radiotracer te zien is op de SPECT-beelden, dan kunt u de graad van de tracerstapeling beoordelen met semikwantitatieve sorteringen. En die traceerstapeling vergelijkt u uiteindelijk met de traceropname in de ribben (zie onderstaande figuur), volgens graad 0 tot en met 3:11
- Graad 0: geen cardiale opname van de tracer en normale opname in de ribben.
- Graad 1: traceropname in het myocard, maar minder dan in de ribben.
- Graad 2: opname in het myocard is gelijk aan de traceropname in de ribben.
- Graad 3: traceropname in het myocard is groter dan opname in de ribben.
In onderstaande figuur ziet u de visuele vergelijking, zowel op planar- als SPECT-beelden.
Is de botscintigrafie negatief of onduidelijk? En is er reden om de patiënt te blijven verdenken van ATTR-CM? Overweeg dan een hartbiopt met congoroodkleuring, immunohistochemie en/of massaspectometrie.11
Wilt u meer informatie over medische beeldvorming en cardiale amyloïdose? Download de samenvatting van de ‘Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis (MECR)'.
De genetische test
Ten slotte is het in sommige gevallen noodzakelijk om de patiënt genetisch te testen en erfelijkheidsadvies te geven. Bijvoorbeeld als u de diagnose ATTR amyloïdose heeft gesteld. Want met genetisch onderzoek kunt u erachter komen of u te maken heeft met erfelijke ATTR amyloïdose, of niet.3
Verdenkt u een patiënt van cardiale amyloïdose? Volg onderstaand diagnostisch algoritme voor de aanbevolen route richting een definitieve diagnose. Het algoritme start op het moment dat de symptomen, ECG, echo, MRI en biomarkers wijzen op cardiale amyloïdose.
Denkt u onderstaand schema vaker te willen raadplegen? Download het diagnostisch algoritme in de vorm van een zakkaartje. Dit kaartje bevat bovendien een overzicht van de belangrijkste symptomen die kunnen wijzen op ATTR-CM.

Cardiale amyloïdose wordt vaak verkeerd gediagnosticeerd. Of het wordt te laat herkend door een laag ziektebewustzijn en andere ziektegerelateerde factoren.13
Ná diagnose ligt de mediane overleving van ATTR-CM tussen de 2 en 6 jaar, afhankelijk van het genotype en de vorm.13,14 Toch is het de bittere realiteit dat die diagnose is vertraagd: gemiddeld met 3 tot 4 jaar.15
Patiënten die lijden aan AL amyloïdose met cardiale betrokkenheid hebben een nóg kortere levensverwachting. Onbehandeld is de overleving niet meer dan 6 maanden.16 Ter vergelijking: patiënten met veel voorkomende vormen van kanker of hartfalen hebben een hogere levensverwachting dan deze patiënten.12
Bij een late diagnose is er sprake van ongehinderde ziekteprogressie. Want het leidt tot uitstel van een passende behandeling. Zonder interventie blijven de amyloïdfibrillen zich opstapelen en loopt de cardiale schade op waardoor de prognose verslechtert. Daarom is een vroegtijdige diagnose essentieel.13
Om dat makkelijker voor u te maken hebben we een overzicht gemaakt van de belangrijkste symptomen die in verband zijn gebracht met cardiale amyloïdose. Inclusief ezelsbrug in de vorm van het acroniem ‘HIDDEN’. In de onderstaande animatie wordt in minder dan een minuut verteld welke symptomen dat zijn.



Bronnen
1. Connors L, et al. Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation. 2016 Jan; 19;133(3):282-90
2. Mohty D, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013 Oct;106(10):528-40
3. Oerlemans, M, et al. Cardiac amyloidosis: the need for early diagnosis. Neth Heart J. 2019;27, 525–536
4. Cyrille N, et al. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol. 2014;114(7):1089-1093
5. Narotsky D, et al. Wild-type transthyretin cardiac amyloidosis: novel insights from advanced imaging. Can J Cardiol. 2016;32(9):1166.e1-1166.e10
6. Rapezzi C, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203-1212
7. Kumar S, at. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30:989–95.
8. Gillmore J, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39:2799–806.
9. Van den Wyngaert T, et al. The EANM practice guidelines for bone scintigraphy. Eur J Nucl Med Mol Imaging. 2016;43:1723–38
10. Gillmore J, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–12
11. Dorbala S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol. doi:10.1007 /s12350-019-01760-6
12. Rapezzi C, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117-124
13. Maurer M, et al. Addressing Common Questions Encountered in the Diagnosis and Management of Cardiac Amyloidosis Circulation. 2017;135:1357–77
14. Papoutsidakis N, et al. Time course of common clinical manifestations in patients with transthyretin cardiac amyloidosis: delay from symptom onset to diagnosis. J Card Fail. 2018 Feb;24(2):131-133 22. Donnelly J, et al. Cardiac amyloidosis: an update on diagnosis and treatment Clin J Med. 2017;84(12 Suppl 3):12–26
15. Arbustini E, et al. Early identification of transthyretin-related hereditary cardiac amyloidosis. J Am Coll Cardiol. 2014;7:511–4
16. Sperry BW, et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol. 2018;72(17):2040-2050
Ook interessant
-
Wim de Ket heeft wildtype ATTR amyloïdose
Eind 2018 kreeg Wim de Ket te horen dat hij ernstig ziek was. De diagnose? Amyloïdose. Wildtype transthyretine amyloïdose met cardiale betrokkenheid, om precies te zijn.
-
Pien heeft AL Amyloïdose
Ongeveer twee jaar geleden werd Pien gediagnosticeerd met AL amyloïdose. Vermoedelijk zijn de klachten járen eerder ontstaan. Maar niemand kan het haar precies vertellen.
-
Dik Schipper over de erfelijkheid van amyloïdose
‘Ik ben nu elf jaar ziek en ben blij dat ik er nog ben. Ik durf niet te ver vooruit te kijken, maar probeer te genieten van de kleine, mooie momenten in het leven.’ Dik Schipper (63 jaar) heeft erfelijke ATTR amyloïdose. Dat werd pas ontdekt toen hij al ernstige klachten had. ‘Gelukkig is het dankzij erfelijkheidsonderzoek bij veel familieleden in een vroeger stadium ontdekt. Zij konden sneller starten met een behandeling.’



