
Amyloïdose cardiologie
Erfelijke ATTR amyloïdose
Als het in de familie zit
Epidemiologie | Symptomen | Diagnose | Erfelijkheidsonderzoek | Ziektemechanisme | Behandeling
Erfelijke amyloïdose kan niet alleen het leven van de patiënt overhoop gooien, maar ook dat van de familieleden. Het is kop of munt: de kans dat deze familieleden de mutatie overerven is 50%.
Op deze pagina kunt u lezen over de epidemiologie, symptomen, diagnose, pathologie en behandeling van erfelijke ATTR amyloïdose (hATTR). Daarnaast gaan we in op het erfelijkheidsonderzoek dat onmisbaar is bij dit type amyloïdose.
Maar eerst: wat is hATTR precies?
Bij ATTR amyloïdose is er een probleem met transthyretine (TTR). Een precursor eiwit dat met name door de lever wordt aangemaakt en verantwoordelijk is voor transport van retinol (vitamine A) en thyroxine (T4). Wat gaat er mis?
TTR is door een mutatie minder stabiel, valt uiteen, de losse stukjes vouwen zich daarna verkeerd op en klonteren samen om ten slotte amyloïdfibrillen te vormen. En, net zoals bij de andere vormen van amyloïdose, infiltreren de fibrillen verschillende organen waardoor die op den duur slechter zullen functioneren.
In het geval van hATTR, ofwel: familiale transthyretine amyloïdose, is TTR minder stabiel door een autosomaal dominante, erfelijke puntmutatie van het TTR-gen. Van de meer dan 100 verschillende mutaties die zijn geïdentificeerd, komt de zogeheten V50M-mutatie het meest voor. Door deze mutatie neemt het aminozuur methionine de plek in van valine op positie 30/50 van TTR.1
Genetisch testen is de manier om deze ziekte op te sporen.
De epidemiologie van erfelijke ATTR amyloïdose
hATTR amyloïdose is zeldzaam. De geschatte incidentie is 3 per jaar (100 patiënten in 33 jaar, van 1985 tot 2018). En de prevalentie is ongeveer 3 per miljoen.2 Hoe ziet dat eruit in de praktijk?
Er staan circa 50 patiënten onder controle van een behandelaar op een totaal aantal van 17 miljoen mensen. Mannen en vrouwen lopen evenveel risico om deze ziekte te ontwikkelen.2
In bepaalde landen komt hATTR met polyneuropathische betrokkenheid (hATTR-PN) relatief vaak voor. Zoals in Portugal, Zweden, Cyprus, Mallorca en Japan. Ter illustratie: in het noorden van Portugal heeft naar schatting 1 op de 538 mensen deze ziekte. Een ander voorbeeld, maar dan niet locatiegebonden: hATTR met cardiale betrokkenheid (hATTR-CM) komt betrekkelijk vaak voor onder de Afro-Amerikaanse bevolking.1,3
hATTR is een onomkeerbare, progressieve ziekte die autosomaal dominant overerft. Dus de ziekte blijft binnen de familie. Dat zorgt ervoor dat hATTR zich op bepaalde plekken concentreert, afhankelijk van waar de getroffen families verblijven. Alhoewel, dan gaan we ervan uit dat familieleden hun hele leven bij elkaar in de buurt blijven wonen. De realiteit is anders.
Migratie leidt ertoe dat familieleden (met hATTR) zich afzonderlijk over de wereld verspreiden. Daardoor is de ziekte mondiaal. Dus verdenkt u een patiënt van hATTR? Dan kan een grondige familiale anamnese u helpen bij het stellen van de diagnose.4,5
De symptomen van erfelijke ATTR amyloïdose1
Hierboven las u al over hATTR-PN en hATTR-CM. Dat zijn de 2 fenotypes die u vooral kunt tegenkomen in de klinisch praktijk. Maar een gemengd fenotype is ook mogelijk.
Bij hATTR-CM hebben de amyloïdfibrillen het hart geïnfiltreerd met cardiale symptomen als gevolg. En bij hATTR-PN is er vorming van amyloïd in de zenuwen waardoor sensomotorische disfuncties ontstaan.
Voor de duidelijkheid: er wordt grofweg onderscheid gemaakt tussen patiënten met ATTR met polyneuropathische betrokkenheid en ATTR met cardiale betrokkenheid.5 Maar het zijn niet twee verschillende aandoeningen; het zijn andere uitingen van dezelfde ziekte.
ATTR amyloïdose is een progressieve systemische ziekte, en de klachten hoeven zich niet te beperken tot het hart en het zenuwstelsel. De amyloïdfibrillen kunnen in diverse organen en weefsels neerslaan waardoor het klachtenpatroon per patiënt zal verschillen.
Hieronder ziet u het volledige scala aan klinische symptomen.
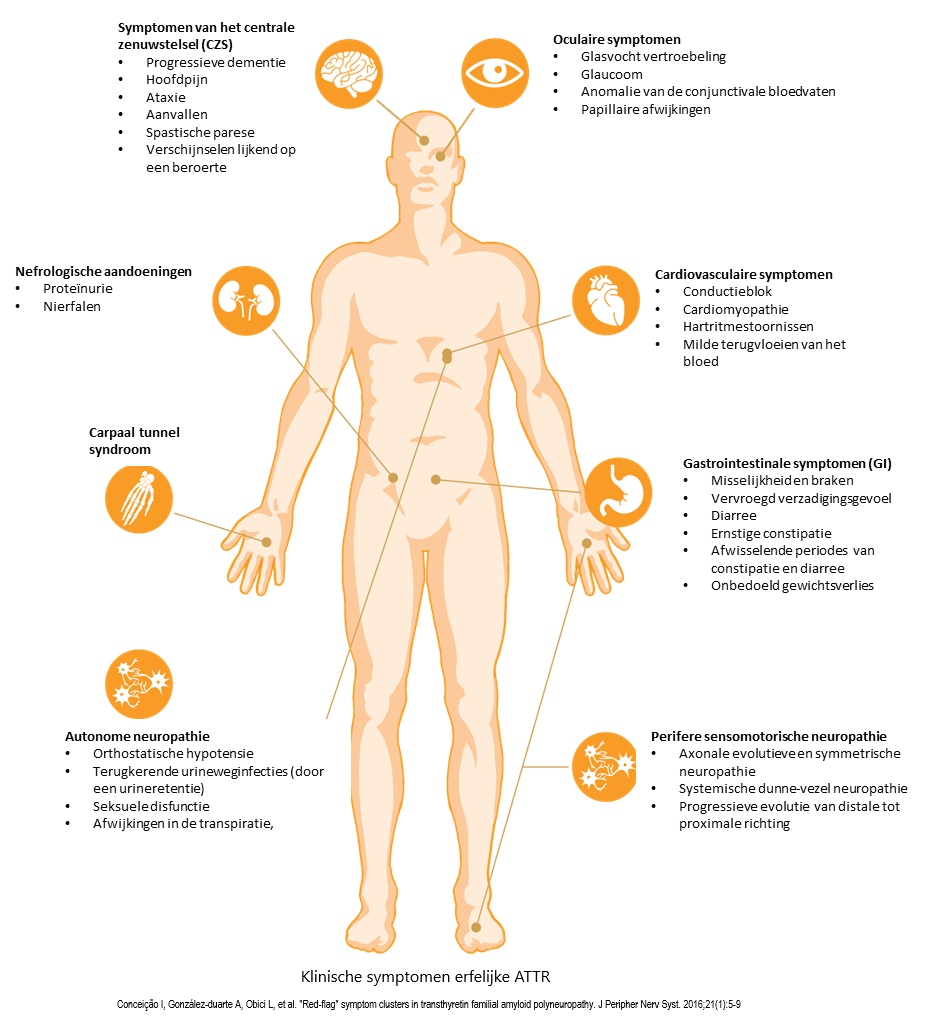
Welke symptomen zou u als eerst kunnen waarnemen bij een patiënt die hATTR heeft? In het begin ontwikkelen deze patiënten autonome klachten; nog voordat ze slechter gaan lopen en hun levenskwaliteit verder achteruitgaat.5
U kunt daarbij denken aan:
- carpaletunnelsyndroom
- tintelingen, gevoelloosheid, pijn en spierzwakte in armen en benen
- afwisselend constipatie en diarree
- vermoeidheid
- slechter zien door vlekken in het gezichtsveld
- gewichtsverlies
- misselijkheid
- erectiestoornissen.
Met welke van deze symptomen de patiënt zich in het beginstadium presenteert, dat kan afhangen van het type mutatie. In een later stadium kunnen hier andere autonome stoornissen bij komen, zoals urineproblemen (bv. urineretentie of incontinentie) en orthostatische hypotensie.1
Het belang van een vroege diagnose
Dag in dag uit wakker worden met klachten die niemand kan verklaren. Van ‘diagnose onbekend’ naar een verkeerde diagnose. En weer terug. Zo leven sommige mensen met ongediagnosticeerde hATTR jarenlang.
Ondertussen tikt de tijd door. Waarvan er niet veel meer over is: de gemiddelde overleving van hATTR-patiënten is 10 jaar, een prognose die verslechtert wanneer er ook sprake is van cardiale klachten.6
Als de diagnose vroeg was gesteld bij deze mensen, dan had die tijd beter benut kunnen worden. Na een diagnose zou het mogelijk zijn geweest om deze patiënten te behandelen; in de hoop hun ziekte enigszins te stabiliseren. Maar het brede spectrum aan symptomen maakt een (vroege) diagnose lastig.
Hoe mooi zou het zijn als patiënten met hATTR er eerder achterkomen dat ze deze ziekte hebben? En wat zou dat betekenen voor hun levensverwachting?
De diagnose van erfelijke ATTR amyloïdose
Waarom is het belangrijk om deze ziekte zo vroeg mogelijk op te sporen? Dat geeft u en uw collega’s de kans om een passende, multidisciplinaire behandeling in te zetten. Een behandeling die de ziekte idealiter stabiliseert en de progressie ervan vertraagt.2 Het verwachte resultaat?
Een verbeterde overleving en lagere ziektelast. Bovendien is een diagnose van hATTR niet alleen waardevol voor de patiënt, maar ook voor de familieleden.2
We noemden het hierboven al: de multidisciplinaire aanpak van de behandeling. Die multidisciplinaire betrokkenheid hoeft zich niet tot de behandeling te beperken en kan al eerder nuttig zijn. Inderdaad, tijdens het diagnostisch traject.2
Multidisciplinaire diagnostiek2
Verschillende specialismen kunnen een rol spelen bij het identificeren van patiënten met hATTR.
Neurologen en pijnspecialisten kunnen deze rol bijvoorbeeld vervullen bij patiënten met tekenen van een beginnende polyneuropathie of autonome neuropathie die niet direct kunnen worden verklaard. Hierbij moet de drempel laag liggen om (naast vrije lichte ketens, NT-proBNP en urineanalyse) via de genetica DNA-diagnostiek te overwegen.
Vervolgens de cardiologie. Stel, een patiënt heeft een verdikt myocard en lijdt aan hartfalen. Maar wat is de exacte oorzaak? De bovenstaande diagnostiek, gevolgd door skeletscintigrafie kan de cardioloog helpen bij deze onverklaarde klachten. Wellicht is er sprake van een verhoogde traceropname in het hart.
Bij de interne geneeskunde en het maagdarmspecialisme zullen regelmatig patiënten langskomen met onbegrepen klachten, zoals diarree en gewichtsverlies. Blijkt er een reden te zijn om een van die patiënten te verdenken van amyloïdose? Dan zijn de hierboven genoemde diagnostische onderzoeken op zijn plaats. Maar ook endoscopie met weefselonderzoek. Congoroodkleuring is daarbij noodzakelijk om de aanwezigheid van amyloïd aan te kunnen tonen.2
Ten slotte zijn we op de afdeling oogheelkunde, waar patiënten zich kunnen presenteren met visusproblemen door glasvochttroebelingen. Dit is ook een aanwijzing voor de vorming van amyloïd.
Bent u op zoek naar de oorzaak van hartfalen bij uw patiënt? Of heeft u een patiënt met onverklaarbare neurologische klachten? Overweeg de mogelijkheid dat hATTR de veroorzaker is en benut de onderstaande tools om duidelijkheid te krijgen. Misschien helpt het u om een diagnose te stellen en de onverklaarbare klachten van uw patiënt beter te begrijpen.
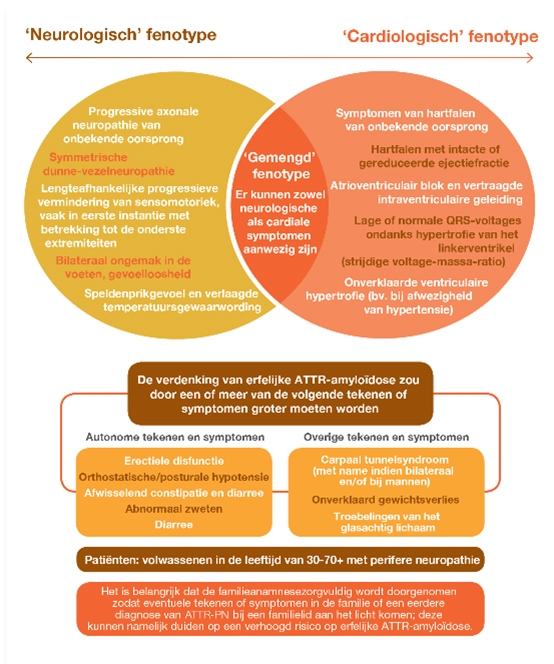
Diagnostisch algoritme Cardiologische symptomen bij ATTR amyloïdose
Diagnostisch algoritme Neurologische symptomen bij ATTR amyloïdose
Op deze pagina gaat het over erfelijke ATTR. Dus we hebben een onmisbaar onderdeel van de diagnostiek nog niet benoemd: erfelijkheidsonderzoek. Alleen met de resultaten van een genetisch onderzoek kunt u, ongeacht uw medisch specialisme, ontdekken of u te maken heeft met hATTR.2
Erfelijkheidsonderzoek
Op de pagina over genetisch testen bij erfelijke ATTR amyloïdose staat het ook: u kunt een doorslaggevende rol spelen in de levensloop van patiënten, en hun familie, bij een verdenking van erfelijke ATTR-amyloïdose.
In welk opzicht? Door ervoor te zorgen dat elke patiënt met deze diagnose in beeld komt bij een klinisch geneticus. Dat is van belang voor de patiënt, maar ook voor eerstegraads familieleden. Want zij hebben 50% (autosomaal dominant) kans dat ze de genmutatie hebben geërfd.1
Meer weten over het belang van genetisch testen en counseling? Luister de Podcast met Dorien Müller, bestuurslid van patiëntenorganisatie Stichting Amyloïdose Nederland.
Genmutatie aangetoond? Verwijs door naar de klinisch geneticus
De klinisch geneticus is dé deskundige als het neerkomt op erfelijkheidsadvies (genetisch counseling) en familieonderzoek.
Uw collega’s die werken bij het Expertisecentrum Amyloïdose (UMCG) adviseren nadrukkelijk om betrokken familieleden (ook wel adviesvragers genoemd) door te verwijzen naar een klinisch geneticus bij hen in de buurt. Zodat de aanwezigheid van een genmutatie kan worden uitgesloten of vastgesteld.2
Indien er sprake is van een genmutatie is onderstenuning vanuit de klinisch genetische afdeling
aanwezig voor de adviesvrager en eventueel betrokken familie. De gendrager kan zelfs beroep doen op preconceptieadvies en krijgt de kans om in-vitrofertilisatie met preïmplantatiediagnostiek en embryoselectie te overwegen.2
Over de interactie met adviesvragers leest u meer in de Richtlijn informeren van familieleden bij erfelijke aandoeningen. Hoort u het liever van de expert zelf? Luister de podcast met Dr. Paul van der Zwaag, klinisch geneticus bij het UMCG.
Bij hATTR draait het aan de ene kant om het opsporen van symptomatische dragers van de genmutatie. In de regel melden deze dragers zich bij een van de hierboven beschreven specialistische afdelingen, nadat ze zijn doorverwezen door de huisarts.
Aan de andere kant gaat het om de adviesvrager. Ofwel: de familieleden die via een familiebrief te horen krijgen dat ze 50% kans hebben drager te zijn van een genmutatie waardoor ziekteverschijnselen van hATTR zich na verloop van tijd kunnen openbaren.
Hoe dan ook, in beide gevallen is tijdige detectie essentieel. In de laatstgenoemde situatie staat het familieonderzoek en het informeren van die familieleden voorop. Van eerstegraads familie tot verre verwanten.
Dus de informatie dat er een erfelijke ziekte in de familie is, moet niet alleen ouders, broers en zussen bereiken, maar ook (achter)neven en (achter)nichten. En zelfs familieleden die zich nog verder in de bloedlijn bevinden.
Toch is dit niet altijd haalbaar. Hoe ver mag de klinisch geneticus gaan in die zoektocht naar verre familieleden? En wat is voor de familie acceptabel? Helaas is dit een grijs gebied waar privacy en bevoegdheden van genetica en familie dwars door elkaar heen lopen. Wellicht kan hier een rol voor de patiënten, vertegenwoordigd via de SAN (Stichting Amyloïdose Nederland), weggelegd zijn.2
We sluiten dit onderdeel af met een talkshow. In onderstaande video ziet u vakgenoten van gedachten wisselen over ATTR amyloïdose en erfelijkheid. Duur: 7 minuten.
Het ziektemechanisme van erfelijke ATTR amyloïdose7,8
In dit onderdeel leest u kort hoe (h)ATTR een patiënt ziek maakt. Om dat uit te leggen beginnen we bij het precursor eiwit transthyretine (TTR) waarna we het ziektemechanisme stapsgewijs beschrijven:
- TTR wordt hoofdzakelijk in de lever aangemaakt en circuleert gewoonlijk als een tetrameer van vier gevouwen eiwitsubeenheden (monomeren).
- Mutaties in het TTR-gen zijn aangetoond als oorzaak voor ATTR polyneuropathie.
- Er zijn meer dan 100 verschillende mutaties van het TTR-gen gerapporteerd. De meest voorkomende mutatie is V50M (voorheen ook genoemd V30M).
- Mutaties veranderen de volgorde van aminozuren en produceren zo monomeren die de vrije tetrameer destabiliseren.
- Destabilisatie resulteert in dissociatie van het tetrameer in gevouwen monomeren, die vervolgens verkeerd vouwen en aggregeren wat uiteindelijk cumuleert in het ontstaan van amyloïdfibrillen.
- Amyloïdfibrillen slaan neer in perifere en autonome zenuwen en in andere organen, zoals het hart, ogen, maagdarmkanaal en nieren.
Visueel ingesteld? Bekijk de onderstaande animatie waarin de pathologie van ATTR amyloïdose in nog geen 2,5 minuut wordt toegelicht.
De behandeling van erfelijke ATTR amyloïdose9
De strategie bij de behandeling van (erfelijke) ATTR is globaal in tweeën te splitsen.
Ten eerste bestaat de therapie uit het behandelen van de aangedane, disfunctionerende organen en de klachten die daar het gevolg van zijn. Een multidisciplinaire aanpak is daarbij onmisbaar. Ook fysiotherapie, diëtiek, psychologie en maatschappelijk werk kunnen onderdeel uitmaken van de behandeling. Dat verschilt per patiënt.
Ten tweede is het mogelijk om het ziekteproces direct aan te pakken met farmacotherapie, afhankelijk van het type amyloïdose en het stadium waarin de ziekte zich bevindt. Met deze farmacotherapeutische behandeloptie kunnen de verschillende vormen van (h)ATTR worden gestabiliseerd waardoor er minder snel amyloïdfibrillen ontstaan.
Voor de erfelijke vorm van ATTR amyloïdose bestaat nog een behandeling: gene silencing. Voor de patiënten die polyneuropathie als klacht hebben, kan dit ook een behandeloptie zijn.
Wilt u meer weten over de behandeling van amyloïdose? Ga naar de website van het Expertisecentrum Amyloïdose (onderdeel van het UMCG). Of neem direct contact op met dit expertisecentrum of dat van het UMC Utrecht.


Bronnen
1. Adams D. et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol 2016, 29 (suppl 1):S14 - S26.
2. Richtlijn diagnostiek en behandeling van erfelijke ATTR amyloïdose GRaCE. https://www.amyloid.nl/wp-content/uploads/2019/12/Richtlijn-ATTRm-amyloidose-versie-3.pdf
3. Maurer M. et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161-172.
4. Swiecicki P. et al. Hereditary ATTR amyloidosis: a single-institution experience with 266 patients. Amyloid. 2015;22(2):123-131.
5. Gonzalez-Duarte A. Autonomic involvement in hereditary transthyretin amyloidosis (hATTR amyloidosis). Clin Auton Res. 2019;29(2):245-251.
6. Stichting Amyloïdose Nederland (SAN), de Vereniging Samenwerkende Ouder- en Patiëntenorganisaties (VSOP) en het Nederlands Huisartsen Genootschap (NHG). Informatie voor de huisarts over amyloïdose. Soest 2016.
7. Benson M. et al. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36:411-423.
8. Hou X. et al. Transthyretin and familial amyloidotic polyneuropathy: recent progress in understanding the molecular mechanism of neurodegeneration. FEBS J. 2007;274:1637-1650
9. Nativi-Nicolau J. et al. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol. 2018 Sep;33(5):571-579
Ook interessant
-
Wim de Ket heeft wildtype ATTR amyloïdose
Eind 2018 kreeg Wim de Ket te horen dat hij ernstig ziek was. De diagnose? Amyloïdose. Wildtype transthyretine amyloïdose met cardiale betrokkenheid, om precies te zijn.


